



Recently, the AIDD team at Viva Biotech published their novel work in the Journal of Chemical Information and Modeling (JCIM) under the title “Pep2MARS: Automated Cyclic Peptide Parameterization for Molecular Dynamics and Compound Design”. This paper was led by Dr. Yue Qian, Vice President and Head of the MARS (Multi-modality AI-rooted Solutions) at Viva Biotech (Shanghai), with Dr. Junhao Li, Project Manager at Viva Biotech, as the first author.

(Source: American Chemical Society Official Website)
Pep2MARS is a Viva Biotech developed platform focusing on peptide modality using multiple AI rooted solutions, which combine both physics-based simulations and wet-lab evaluations. The current work addresses the increasing computational complexity in physics-based modeling of various cyclic peptides. The following section presents further elaboration on the methodology.
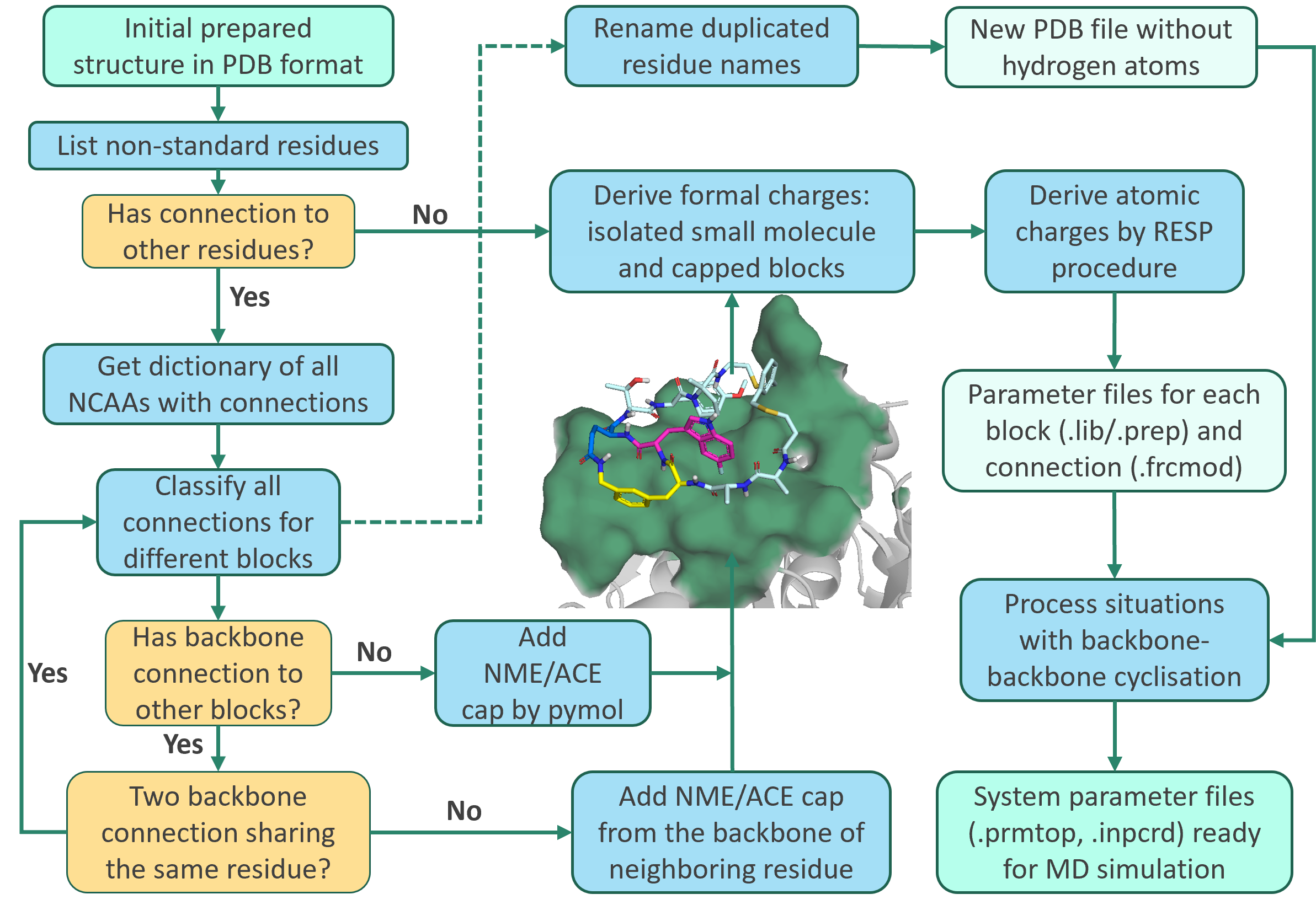
For cyclic peptides containing non-canonical amino acids, complex side-chain crosslinks, and multi-ring scaffolds, parameterization prior to molecular dynamics (MD) simulation remains highly challenging. Tasks such as residue naming standardization, covalent bond assignment, charge redistribution, and force-field mapping are highly error-prone. Under the widely used Amber force field framework, in particular, the presence of non-standard residues and diverse cyclization patterns often renders manual parameterization both time-consuming and susceptible to hidden errors.
The team developed an automated parameterization workflow designed for complex cyclic peptide systems. With a clear goal: starting from a three-dimensional structure—either derived from experimental design or wet-lab data—it can automatically identify non-standard residues and covalent connectivity patterns and generate topology and parameter files compatible with the Amber force field. This transforms cyclic peptide MD preprocessing from a labor-intensive, expertise-dependent manual task into a standardized, reproducible, and scalable computational workflow.
A key limitation of traditional single-residue parameterization approaches is their tendency to force cyclic peptides into linear peptide modeling assumptions, thereby neglecting the chemical coupling effects introduced by cyclization in charge assignment. Pep2MARS addresses this issue through a “block-based modeling” strategy, which systematically partitions molecular structures into different blocks according to bonding patterns. This unified representation enables consistent fitting of atomic charges and force-field parameters for complex peptides involving various linkages, as well as diverse cyclization strategies. This approach transforms the parameterization process for complex cyclic peptide molecular dynamics from an experience-dependent, manual procedure into an automated workflow that is governed by chemically defined rules and is both verifiable and reproducible. The workflow has been validated on ten structurally complex PCSK9 cyclic peptide inhibitor systems. It demonstrated robust automated recognition of cyclization patterns and accurate generation of simulation-ready parameter files. Moreover, molecular dynamics trajectories generated using these parameters align well with published results obtained from alternative commercial software platforms.
(Source: This study, reused with permission from CCC. Copyright © 2026 American Chemical Society)
Overall, this workflow not only resolves the automation bottleneck in cyclic peptide parameterization, but also integrates structural modeling, parameter generation, and molecular dynamics interpretation into a unified and reusable computational framework, providing a methodological foundation for cyclic peptide drug design. From a broader perspective in biomedical R&D, the innovation lies in transforming cyclic peptide simulation preparation from a niche, expert-dependent procedure into a scalable capability accessible to broader research teams. Particularly in today's landscape—where AI-driven peptide design, structure prediction, virtual screening, and experimental validation are accelerating and coupling together—molecular dynamics simulation increasingly acts as a critical mechanistic sieve bridging “design” and “validation.” The ability to stabilize and standardize this step will be a key determinant in achieving closed-loop design for complex molecular systems.
The workflow has now been deployed as a dedicated module within Viva Biotech's AIDD platform, primarily supporting internal users in more efficient parameterization of complex cyclic peptides and related systems. This critical advancement directly influences the reliability of downstream simulations and the overall efficiency of the drug discovery pipeline.
For further details on the study, please refer to the full paper (subscription access to ACS is needed):
Li, J. and Qian, Y. (2026). Pep2MARS: Automated Cyclic Peptide Parameterization for Molecular Dynamics and Compound Design. Journal of Chemical Information and Modeling. doi:https://doi.org/10.1021/acs.jcim.6c00340.