



Recently, AIxplorerBio, a Viva Biotech portfolio company, published an article titled "Adaptive lambda schemes for efficient relative binding free energy calculation" in the Journal of Computational Chemistry. Dr. Yue Qian, Executive Director of Computational Chemistry at Viva Biotech, is the corresponding author of this paper. This is a highly productive collaboration between AIxplorerBio and Dr. Qian.

Source: Journal of Computational Chemistry
The research represents a significant enhancement to the FEP platform, introducing adaptive lambda schemes for the effective calculation of relative binding free energy. The main focus of the research includes:
The relative free energy perturbation (RFEP) calculation is one of the most theoretically sound computational chemistry approaches for the binding affinity prediction. However, its application is often hindered by the complexity of the calculation choices and the requirement of expertise in the field. Improper lambda scheme of RFEP may result in deviations from an accurate description of the perturbation process and is prone to erroneous affinity predictions. To address such challenges, an automated adaptive lambda method is proposed where the adaptive lambda schemes are obtained through a split-and-merge algorithm based on the pilot runs. The newly established workflow along with a series of improvements to the perturbation settings increases the consistency of the RFEP calculation results. Comparing the pilot and adaptive lambda schemes, the latter demonstrated improvements in convergence and reproducibility and lowered the mean unsigned error and the root-mean-square error. Overall, the adaptive lambda method is a reliable and robust choice to predict small molecule relative binding free energy and can be capitalized to benefit routine RFEP calculations for drug discovery projects.
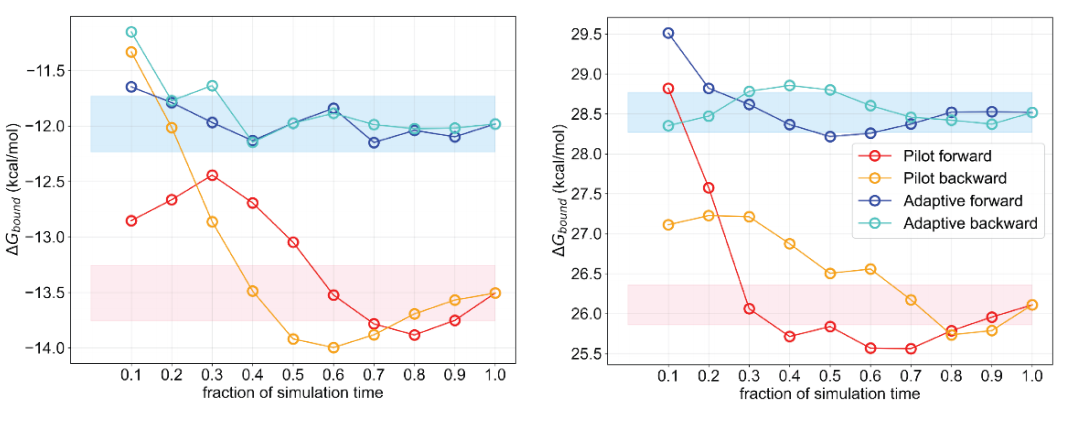
Source: Adaptive Lambda Schemes for Efficient Relative Binding Free Energy Calculation
The research findings have been effectively integrated into AIxplorerBio's small molecule dual-target drug design platform, AIxDDDTM. This platform encompasses three technical modules: target combination assessment, molecular design, and optimization. It aids in the rapid theoretical validation of the synergistic effects of target combinations and the design and optimization of small molecule drugs with high activity and specificity for two targets simultaneously.
The target combination assessment module of AIxDDDTM. leverages the latest advancements in natural language processing (NLP) and large language model (LLM) to analyze relevant multi-omics data. It scores and ranks potential synergistic effects (additive efficacy, antagonistic toxicity) of target combinations in specific disease areas. The molecular design module, inspired by the thought processes of medicinal chemistry experts, utilizes AI technology to generate and continuously refine molecules based on pharmacophore-target interactions, employing reinforcement learning. AIxplorerBio's proprietary free energy calculation method, including the results from this article, plays a crucial role in compound optimization, assisting in the GO/NO GO decision-making process prior to the synthesis of newly designed compounds.
As an expert in the CADD field, Dr. Yue Qian possesses extensive knowledge and practical experience in computational chemistry. As the corresponding author of this paper, she contributed the research ideas, provided professional guidance, and led the writing of the research article.
At Viva Biotech, she leads a professional team that has independently developed an FEP platform from scratch, utilizing the company's high-performance computing system. This platform integrates a user-friendly interface, automated analysis, and golden-standard precision. As this platform is internally developed at Viva Biotech, our research scientists have a comprehensive understanding of the methods and can fine-tune the parameters to optimize FEP calculation conditions with first-hand experiences. Viva Biotech's proprietary free energy perturbation method shows a small mean unsigned error (MUE) of 1 kcal/mol, indicating a high level of accuracy. With the support of the high-performance computing clusters, FEP calculations have now become a routine practice for most of the drug discovery services provided by Viva Biotech.
For further details on the paper, please refer to: J. Zeng, Y. Qian, J. Comput. Chem. 2023, 1. https://doi.org/10.1002/jcc.27287
About AIxplorerBio
AIxplorerBio is an AI-powered drug R&D biotech company. We focus on the discovery and development of new medicines for immunological and neurodegenerative diseases. We are also committed to creating a new AI-powered drug R&D paradigm to enable innovation, efficiency and precision in searching for better medicines.
AIxplorerBio is established by a team of highly experienced cross-discipline experts and sponsored by the new B/IT (biotechnology and information technology) powerhouse, BioMap, and the leading drug discovery CRO platform, Viva Biotech. In addition to the strategic cooperation with BioMap and Viva, AIxplorerBio also actively seeks collaboration opportunities with AI-oriented technology companies as well as drug R&D companies to explore more efficient ways for new drug R&D.