



Home / News / Events / Events detail
Recently, Viva Biotech successfully hosted a Viva BioInsights Webinar, centered on the theme "Innovative Pathways in Drug Discovery: Integrating DNA-Encoded Library (DEL) Technology". The event featured Dr. Bing Xia, Vice President of Viva Biotech, and Dr. Wes Kazmierski, a Medicinal Chemist as speakers. They provided an in-depth exploration of DEL technology, starting from the concept of DEL technology, particularly emphasizing how DEL technology accelerates the R&D process. This was illustrated through the case study of identifying a Best in Class (BIC) IDO1 inhibitor. In the article below, we have compiled the highlights of this webinar. For those interested in viewing the session, you can click the provided link to watch the replay or visit our YouTube Channel for more insightful content.
Dr. Bing Xia 1: In 1992, Nobel laureates in Physiology and Medicine Sydney Brenner, and Richard Lerner, first proposed the concept of DNA Encoded Library (DEL), but the emergence of the second-generation sequencing in 2005 made DEL a more economically feasible approach. Since then, DEL technology has evolved significantly and become a key component in drug discovery. The workflow of DEL involves several fundamental stages: construction of DEL libraries, affinity selection, NGS sequencing, data analysis, off-DNA re-synthesis, and hit evaluation. DEL technology offers many revolutionary benefits: the “Split & Pool” strategy of library synthesis can exponentially expand the diversity of DELs; affinity selection minimizes the need for upfront assay development, requiring only a minimal amount of protein and knowledge of the target of interest, which makes DEL perfect for First-In-Class (FIC) targets. DEL platform reduces cycle time and operation costs as well. However, we highly recommend synergies between DEL screening and other hit ID methods.
Our V-DEL platform is differential and fine-tuned to this holistic approach:
1) We are the top branded CRO specializing in protein science worldwide, with the largest operational size in protein production (including membrane proteins) and structural biology;
2) Viva Biotech constructs high quality V-DELs using novel on-DNA reactions and strategies; V-DEL uses lots of legacy building blocks from Viva’s med chem engine, which makes our V-DELs unique. With substantial support from those med chemists (>50), V-DEL significantly shortens both the timelines for customized DEL synthesis and off-DNA re-synthesis for hit confirmation--the current bottleneck of this powerful technology;
3) Viva Biotech stands out as one of the most experienced providers in the field for ASMS and SPR validation of DEL hits;
4) Viva Biotech has developed and validated a range of synergetic approaches, such as (DEL+FBDD), (DEL+FEP calculation), to tackle otherwise challenging projects that might be difficult when using only one of those platforms;
5) Viva Biotech’s high-caliber CADD + AI group leveraging its powerful computational infrastructure and GPU clusters can enhance the outcomes of DEL screening and its follow-up investigations.
6) While you work with one platform at Viva, our entire team of experts with rich experience spanning nearly 20 platforms -- including protein production, ASMS, SPR, X-ray crystallography, Cryo-EM, FBDD, SBDD, all the way to medicinal chemistry, ADME-PK, CADD, AIDD and CDMO -- is always accessible.
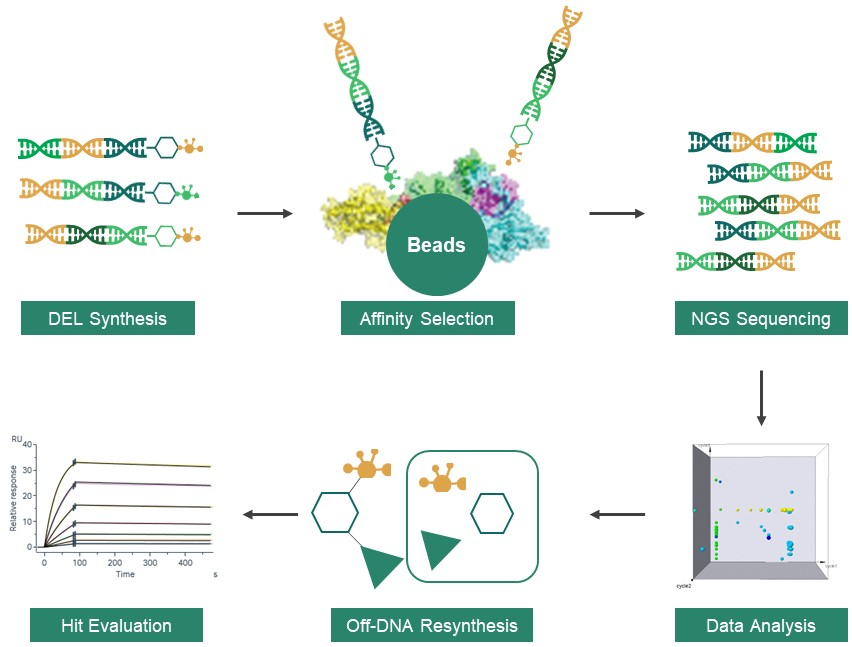
Scheme 1 1: Workflow of V-DEL
Dr. Wes Kazmierski 1: First, I'd like to introduce two articles that were published in “Medicinal Chemistry” and “ChemBioChem”. The papers, titled "DNA-Encoded Library Technology-Based Discovery, Lead Optimization, and Prodrug Strategy toward Structurally Unique Indoleamine 2,3-Dioxygenase-1 (IDO1) Inhibitors" and "Characterization of Apo-Form Selective Inhibition of Indoleamine 2,3-Dioxygenase", were co-authored by me and my colleagues. Dr. Xia also made a key contribution to the first article. These papers provide additional details, beyond what is presented in the Webinar. IDO1 is a heme-containing enzyme that catalyzes the oxidation of tryptophan to N-formyl kynurenine, the first and rate-limiting step in the Kynurenine Pathway. IDO1, along with its closely related isoforms IDO2 and TDO, collectively accounts for over 90% of total tryptophan catabolism in mammals. IDO1 is induced or upregulated in response to pathogen infection or cellular inflammation, limiting T-cell function and leading to immunosuppression and immune tolerance onsite. IDO1 facilitates cancer cells escape detection by the immune system and thus IDO1 inhibition was thought to cause anti-cancer outcome. Depletion of tryptophan and overproduction of kynurenine (which can be cumulatively expressed as a high K/T ratio) has also been linked to CNS diseases, such as Alzheimer's, as well as increased morbidity and mortality in HIV-suppressed patients on antiretroviral therapy. Not surprisingly, IDO1 inhibition had been a very crowded and competitive area of pharmaceutical research and development. Many initial clinical IDO1 inhibitors were characterized by less than desirable potency and pharmacokinetic properties, requiring high and multiple daily doses, which was less than ideal in terms of supporting patient’s drug compliance. Therefore, we aimed to discover new chemotypes to address this issue through both High Throughput Screening (HTS) and DEL screening methods.

(Left image is sourced from: J. Med. Chem. 2020, 63, 7, 3552–3562, Publication Date: February 19, 2020, https://doi.org/10.1021/acs.jmedchem.9b01799 ; Right image is sourced from:Chembiochem. 2021 Feb 2;22(3):516-522. doi: 10.1002/cbic.202000298. Epub 2020 Nov 16. PMID: 32974990. )
Dr. Bing Xia 2: A “starting point” or even a probe is highly desirable in the development of FIC drugs, and novel chemotypes are often required during the evolution of BIC drugs. DEL screening can play a significant role in both scenarios. Here are some tech details in our case of DEL screening against IDO1: we used a compound in its Phase II clinical trial as a competitor and included ascorbic acid and methylene blue in the selection buffer to ensure a proper redox system. It's important to note that the screening temperature was set at a nonconventional 37°C. To our excitement, the preliminary screening results suggested that IDO1 is a very DEL-friendly target. During the data analysis process, we preferred chemotypes with signal strength greater than 4.01 and the signal intensities decreased with the presence of the known active site inhibitor, demonstrating clear evidence of SAR (both internally and across libraries), having MW under 450 and cLogP smaller than 4, for off-DNA re-synthesis. Nine chemotypes meeting these criteria underwent Post Selection Chemistry (PSC) and, to our delight, seven of them were confirmed in cell assays! Our prioritizing DEL76* series was based on a deep understanding of the target: this chemotype does not have the highest enrichment value, but it features an indole core similar to tryptophan, the enzyme’s natural substrate. Despite confirmation in cell assays, we observed a puzzling disconnection with biochemical assays. Upon closer examination, we realized that both the DEL selection and cell-based assay were conducted at 37 °C, whereas the HTS enzyme assay was performed at room temperature (25 °C). What a difference 12 degrees makes! But why?
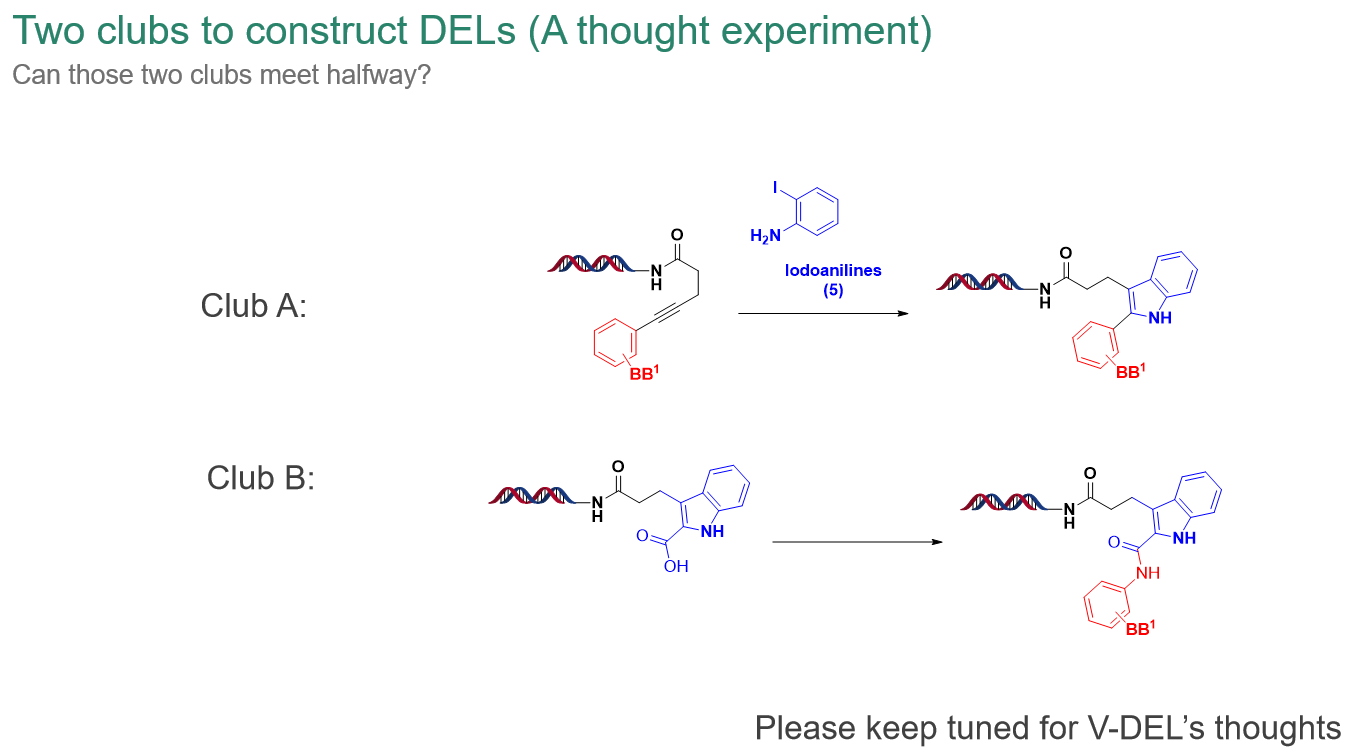
*After Dr. Xia shared the details of synthesizing DEL 76 DEL library using Larock heterocyclization, he further introduced the two main approaches in the current industry for assembling DEL libraries (as shown in the slide below): a) directly forming heterocycles on-DNA, and b) attaching heterocycles to DNA first and then decorating them, along with their respective pros and cons. For example, the former produces molecules with better rigidity, while the latter builds DELs with higher diversity. Following that, he proposed new ideas and strategies for V-DEL construction, promising to elaborate on them in the upcoming Webinars. Please stay tuned.
Dr. Wes Kazmierski 2: To unravel this mystery, we pursued a parallel exploration of the mechanism of action of IDO1 using lead compound GSK5628, developed a bit earlier in another, unrelated internal lead optimization effort. The findings from this research, titled "Characterization of Apo-Form Selective Inhibition of Indoleamine 2,3-Dioxygenase," were published in Chembiochem in 2020. Our data suggested that, unlike traditional IDO1 inhibitors like epacadostat, which bind holo-IDO1 (heme-containing), GSK5628 competitively binds apo-IDO1 (heme-less enzyme). The use of this second mechanism requires a longer incubation time (or higher temperature) to account for the additional holo to apo shift (loss of heme), resulting in the initial discrepancies observed in our previous experiments conducted under slightly different temperatures. DEL76 molecules were also shown to inhibit IDO1 by binding apo-IDO1, akin to GSK5628. Having verified the Mode of Action (MoA) of the new DEL hit 1, we launched optimization and structure-activity relationship (SAR) efforts. Our lead 11 ("non-crystalline 11") demonstrated high bioavailability in rats, but when 11 was crystallized, the "crystalline-11" exhibited a drastic bioavailability reduction in both rats and dogs. We determined FaSSIF solubility of crystalline 11 and concluded that this compound’s low bioavailability was due to its low solubility and poor solubility-limiting absorption. One strategy to address this issue was to explore prodrugs of 11. Among a number of prodrugs made and evaluated in vivo, phosphono-oxymethyl 31 and its crystalline Tris salt version 32 proved highly absorbed from oral dosing, yielding a complete and fast conversion to 11 in plasma. The pharmacokinetic properties of 32 and human pharmacokinetics simulations suggested that a 60 mg BID dose of 32 would deliver enough 11 into the systemic circulation to inhibit >90% of IDO1 at any given time over the 24h period. In summary, after identifying the issue of DEL hits not being validated in standard enzyme activity assays, we were ultimately able to successfully utilize DEL hit 1 through a sequential MoA and medicinal chemistry-based SAR and prodrug design. DEL significantly shortened the drug discovery timeline as hit 1 featured some pre-optimization, and required the synthesis of only about 200 analogues in the lead optimization campaign--a small fraction of what a typical med chem project would require at this stage. At the same time, a sophisticated mechanistic understanding and bespoke med chem designs were necessary to utilize DEL hit 1 and develop it into a clinical- level molecule 32, underscoring the need for an integrated approach.
Dr. Bing Xia 3: I’d like to introduce another tale of DEL/hit ID synergy. As we successfully developed the DEL76 spiro ring series and because a backup series is always preferable when lead a med chem campaign, we reviewed the DEL76 selection data in a “cube view”, and noticed there is a neighboring line, which helped us discovering a unique, unusual way to open the DEL 76 sprio ring series once we aligned the chemical structures being represented by these two SAR lines. Further scaffold hopping of this newly obtained spiro ring opening series with a computationally designed scaffold rendered another preclinical candidate for IDO1! DEL played a pivotal role in the IDO1 campaign, as summarized below.
- Identified multiple lead series with diverse chemical structures (80% hit confirmation rate)
- Observed a disconnection between cell-based and biochemical assays, and rescued hits that HTS would miss (data not shown here)
- Yielded compounds with unprecedented MoA – bind IDO1 via competing with heme for apo-IDO1
- Synergy between HTS and DEL– 1) helped program team understand IDO1 better (temperature effect on enzyme activity). 2) open spiro ring series and then DEL/HTS/Computational design hopping
- DNA attachment point facilitated design of probe for pull down experiment (multiple tool compound series, data not shown here)
- Prodrugging and discovery of a crystalline salt elevated series to pre-candidate stage
A take-home-message is: “Do not treat a DEL hit just like any other hits, please place it back into its selection and library context. You will surprise yourself with how much more valuable information you can extract.” As a huge DEL believer and the leader of V-DEL, I want to promote the synergy between DEL screening and other platforms once again. Viva Biotech’s solution follows the first principle: “Affinity first, specificity and potency pave the path, good molecules will come naturally”.
For more detailed information, please feel free to reach out to our expert team via email at info@vivabiotech.com.